NF-κB Signaling Pathway
- On This Page
- NF-κB signaling pathway
- Interactive NF-κB signaling pathway poster
- Our NF-κB signaling antibody range
- References
The transcription factor Nuclear Factor-Kappa B (NF-κB) is a major regulator of inflammation and controls many aspects of innate and adaptive immune responses. However, constitutive activation of NF-κB is associated with chronic inflammatory and autoimmune diseases and cancer, so inhibition of NF-κB signaling is often a therapeutic goal (Gupta et al. 2010).
Instead of being a single protein, NF-κB represents a series of transcription factors which each bind to specific DNA elements and elicit a variety of transcriptional responses (Gilmore et al. 1999). NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and c-Rel are all members of the NF-κB family and share homology through a Rel homology domain (RHD) which allows NF-κB proteins to form dimers with other NF-κB proteins, and these dimers have transcriptional activity (Liu et al. 2017).
Activation of NF-κB can occur through two separate signaling pathways known as canonical (classical) and non-canonical (alternative) signaling. These pathways are induced by different stimuli, involve different NF-κB family members, and elicit different responses. Although NF-κB represents a family of transcription factors, the most commonly referenced heterodimer of NF-κB is p65-p50 and most stimuli that activate NF-κB signal through the canonical pathway. p65-p50 is referred to as “NF-κB” throughout this page, and the description below refers to the canonical signaling pathway.
Because it elicits a response by binding to nuclear DNA, the activity of NF-κB is regulated by its localization within the cell. Under non-inflammatory conditions, NF-κB is bound by inhibitor of kappa B (IκB) proteins in the cytoplasm. IκB blocks the nuclear localization signal (NLS) of NF-κB and prevents NF-κB from translocating to the nucleus. When the cell senses an inflammatory stimulus, such as the cytokine tumor necrosis factor alpha (TNF-α) or bacterial lipopolysaccharide (LPS), the IκB kinase (IKK) complex is activated and phosphorylates IκB, targeting IκB for destruction. The breakdown of IκB by the proteasome releases NF-κB, which travels to the nucleus where it can activate the transcription of hundreds of genes involved with inflammation and innate and adaptive immunity. The pattern of gene expression depends on the context, which appears to allow NF-κB to generate a specific immune response appropriate to the threat (Zambrano et al. 2016).
The linear process leading from IκB degradation to the transcription of NF-κB target genes does not represent the full story of NF-κB signaling. Instead, nuclear NF-κB acts as a biological oscillator by upregulating transcription of its own inhibitors, including IκB alpha (IκBα). Newly-synthesized IκBα translocates to the cytoplasm, where it binds free NF-κB, sequestering it in the cytoplasm. This causes less NF-κB to translocate to the nucleus, therefore lower levels of IκBα synthesis, which in turn results in a greater proportion of free cytoplasmic NF-κB. The result is an oscillatory process during which levels of nuclear NF-κB are high when total IκBα levels are low, and vice versa, representing a negative feedback loop (Figure 1). Multiple other processes contribute to the behavior of NF-κB oscillations, including the protein A20. A20 is a transcriptional target of NF-κB and inhibits IKK directly, establishing a second negative feedback loop (Nelson et al. 2004). There is evidence to suggest that the pattern of NF-κB oscillation influences the pattern of gene expression, and therefore fine-tunes the inflammatory response (Zambrano et al. 2016).
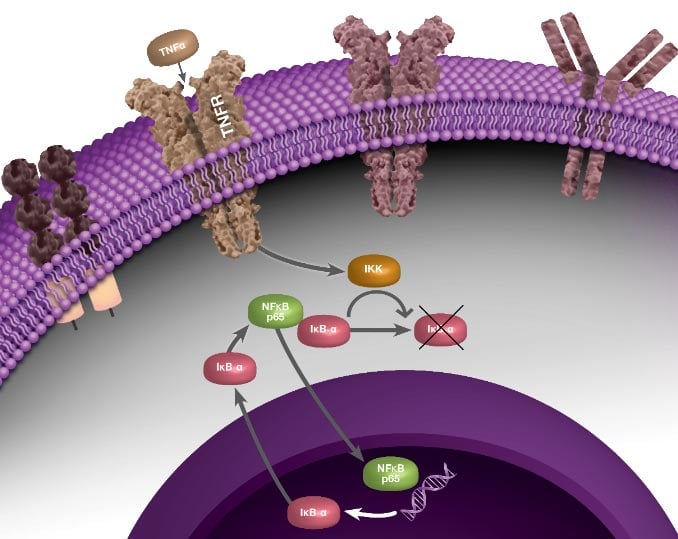
Fig. 1. Negative feedback loop involved with NF-κB signaling, during which NF-κB activates transcription of its own inhibitor, IκBα.
Usually, NF-κB is activated only under certain conditions, which include infection and injury. However constitutive NF-κB activation plays a role in inflammatory and autoimmune disorders and malignancies, and underlies the chronic inflammation involved with inflammatory bowel disease (IBD), rheumatoid arthritis, atherosclerosis, and a variety of malignant tumors (Park and Hong 2016). In fact, NF-κB plays a role in the development, maintenance, and progression of most chronic illnesses, by causing transcription of genes involved with inflammation, cell proliferation and survival, angiogenesis, and tumor promotion and metastasis (Xia et al. 2014).
Many tumor types have constitutively-active NF-κB, which keeps cancer cells proliferating and protects them from apoptosis. In addition, the tumor microenvironment often harbors constitutive NF-κB signaling, which results in accumulation of pro-inflammatory, pro-tumorigenic cytokines, and further maintains a favorable environment for tumor cells. In some cases, inflammatory micro-environments can lead to cancer development. For example, in patients with inflammatory bowel disease (IBD), immune cells in the gastrointestinal mucosa secrete pro-tumorigenic cytokines such as TNF-α, which elevate NF-κB activity. This increases the risk of colon cancer for patients with IBD.
Because of its role in numerous diseases, NF-κB inhibition has become a therapeutic goal. Blocking NF-κB in cancer cells has been observed to prevent proliferation, cause cell death, or make the cells more sensitive to anti-cancer agents. However, because NF-κB is ubiquitously-expressed and critical for human health, inhibition is not straightforward. Many strategies to inhibit NF-κB signaling focus on inhibiting protein kinases and phosphatases involved with the NF-κB pathway, as well as ubiquitination, acetylation, methylation, and DNA binding steps of NF-κB activity, among other processes. Natural and synthetic glucocorticoids, including dexamethasone and prednisone, inhibit NF-κB through inhibition of DNA binding activity and through inhibition of IKK activity. In addition, many nonsteroidal anti-inflammatory drugs (NSAIDs) including aspirin and ibuprofen can inhibit NF-κB in cell culture (Gupta et al. 2010).
Interactive NF-kB Signaling Pathway Poster
We have compiled key proteins involved in the NF-kB signaling pathway into one handy poster you can download for your reference.
Click the pathway components within the images below to discover the key antibodies that Bio-Rad provides.
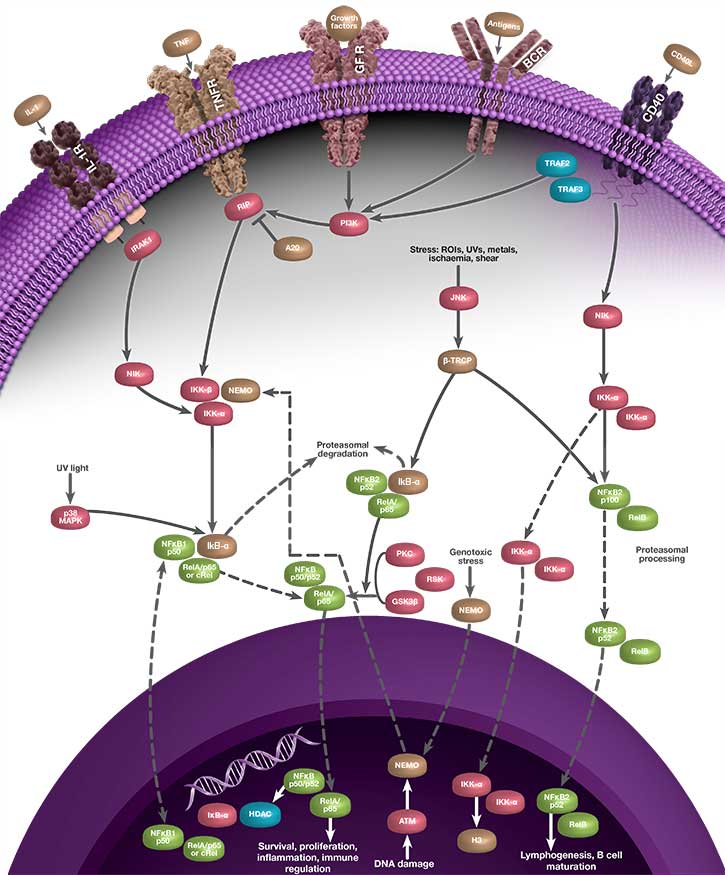
Bio-Rad’s NF-κB Antibody Range
Bio-Rad offers the antibodies needed for your NF-κB research, as well as technical advice to support your experiments.
Browse our range of antibodies against targets involved with NF-κB signaling pathway below. Please use the filters to sort the attributes in the table below in order to find the antibody that fits your exact requirements. If you need any further assistance please do not hesitate to contact us.
Antibodies against targets involved with NF-KB Signaling
| Description | Target | Format | Clone | Applications | Citations | Code |
|---|
- Growth factors
- EGF R antibodies
- Kinases
- Transcription factors
- Translation factors
- Phosphatases
- Phosphatases in cancer
- Phospho-Specific precisionAb antibodies
- Phospho-specific antibodies
- Sumoylation mediated signaling
- Ubiquitination mediated signaling
- Tyrosine phosphorylation of EGF R
- Signal transduction antibodies
References
- Liu T et al. (2017). NF-κB signaling in inflammation. Sig Transduct Target Ther 2, 17023.
- Gilmore TD. (1999). The Rel/NF-kB signal transduction pathway: introduction. Nature 18, 6842–6844.
- Gupta SC et al. (2010). Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochem Biophys Acta 1799, 775–787.
- Nelson DE et al. (2004). Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 306, 704–708.
- Park MH and Hong JT (2016). Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 5, 15.
- Xia Y et al. (2014). NF-κB, an active player in human cancers. Cancer Immunol Res 2, 823–830.
- Zambrano S et al. (2016) NF-κB oscillations translate into functionally related patterns of gene expression. eLife 5, e09100.