Sample Preparation
Cell Lysates
Crude cellular lysates are the most common direct source of starting material used in Western blotting. They can be prepared from immortalized cell lines known to express the target protein, or from transfected cells carrying a protein expression vector. Many different cell types (mammalian, insect, yeast, or bacteria) can be used to supply the protein needed with slight variations in the preparation procedure.
In most cases, the cells are harvested, washed, and lysed to release the target protein. For best results, all these steps should be carried out in a cold room, or on ice. This will minimize proteolysis, dephosphorylation, and denaturation, since all begin to occur once the cells are disrupted . It is possible to simply lyse cells directly in gel loading buffer if a quick check is all that is required. However, sonication may be neccessary to disrupt the highly viscous cellular DNA. Usually, 20-50 mg of cellular lysate is loaded per lane for gel electrophoresis.
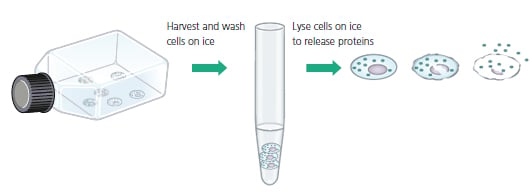
Fig.6. Cells are Harvested, Washed and Lysed to Release Proteins
Choosing the proper lysis buffer and determining an appropriate volume is often a trial and error process that is affected by the type of protein being isolated as well as the particular cells used as a source. Lysis buffers vary from very gentle ones with no detergent to harsher solutions such as rIPA (radio Immuno Precipitation Assay) buffer, which is denaturing and contains multiple detergents.
Typically, NP-40 (Nonidet P-40) lysis buffer, with a milder non-ionic detergent, is used for the isolation of soluble cytoplasmic proteins. At other times, rIPA buffer is chosen because it reduces background, and because sometimes multiple detergents are required to fully release membrane bound or nuclear proteins.
Consideration should also be given to the antibody-antigen interaction which may be affected by the changes to the target protein during lysis.
The amount of lysis buffer is determined based upon a cell count, or else it is estimated based upon the size of the tissue culture vessel. The accompanying table provides some suggested starting points.
Sometimes mechanical disruption, such as with sonication or dounce homogenization, is required to fully release proteins from certain cell and tissue samples. Sonication is also used to break down cellular DNA which can interfere either due to its high viscosity or via non-specific binding. After lysis and centrifugation, the amount of protein in each lysate is measured.
Cell Lysis Volume Recommendations
| Type of cells | Amount of Material | Volume of lysis buffer |
|---|---|---|
| Tissue Culture |
107 cells or 100 mm dish |
1ml |
| Whole Tissue | 100 mg | Add 2 ml and sonicate or dounce homogenize |
| Bacteria | Spin sample, estimate volume | Add 10 volumes and vortex |
| Yeast | Spin sample, estimate volume | Add 10 volumes, then sonicate or vortex with glass beads |
Tissue Samples
Tissue samples display a higher degree of structure than cultured cells and thus may require higher levels of mechanical intervention in order to release the protein of interest. They may also contain multiple cell types which are differentially responsive to the lysis buffer chosen.
Smaller solid tissue samples (up to 100 mg) are placed in ice cold extraction buffer and homogenized on ice, usually with sonication or a douncing rod to facilitate cellular disruption.
Alternatively, and more often with larger tissue samples, a blender is used to homogenize the tissue in PBS, and then cell lysis buffer is added. Once the tissue has been homogenized and lysed, the solubilized cellular components are clarified by centrifugation and tested for protein concentration prior to loading on a gel.
Purified or Semi-purified Extracts
The simplest source of starting material for Western blotting is purified or semi-purified protein samples that are produced in the course of protein purification. These samples rarely require any further manipulation and are simply mixed with gel electrophoresis loading buffer (Laemmli sample buffer).
When using a purified or semi-purified protein preparation, it is possible to load a much smaller amount of total protein onto the gel. Usually 0 .5-1 mg of purified or semi-purified protein is sufficient to observe a strong signal. If unsure, results can be improved by loading several dilutions of the sample.
Determining Protein Concentration
To ensure that samples are in the proper range of detection for the assay, and so they can be compared on an equivalent basis, it is important to know the concentration of total protein in each sample. There are various methods available for determining protein concentration using in-house or commercially supplied kits and reagents.
The simplest method entails measuring the absorbance of the lysate solution at 280 nm or 205 nm. Alternatively, several protein assays are available which rely upon the reduction of metal ions by the peptide bond, e .g. the Lowry and BCA assays; or by dye binding, as with the Bradford assay.
In all instances, a color change results that is proportional to the amount of protein in the sample. Protein concentration is determined by comparison of the target samples to a known standard, such as BSA (Bovine Serum Albumin) diluted in lysis buffer. To get the most accurate measure of protein concentration, it is advisable to test a few dilutions of the sample ensuring that the results lie in the linear range of the protein assay.
Loading Buffer
Once the protein concentration has been determined, samples are diluted in gel loading buffer, also called 2x Laemmli sample buffer. This buffer contains glycerol so that the samples sink easily into the wells of the gel, and a tracking dye (bromophenol blue) which migrates through the gel first to indicate how far the separation has progressed.
For most routine Western blots, SDS (sodium dodecyl sulfate) and a reducing agent are also present in the gel loading/ sample buffer to fully denature the protein and remove all higher order structure. See the accompanying table for the range of loading buffer options and the supplements they contain.
Samples are heated in gel loading/sample buffer for either 5 minutes at 100°C, or 10 minutes at 70°C to aid in the denaturation. At this point, samples can remain at room temperature if they are to be used immediately, or placed at 4°C or -20°C for later analysis.
Sample Buffer Conditions
| Gel Conditions | SDS | DTT or ßME | Comments |
|---|---|---|---|
| Denaturing & Reducing | + | + | Removes all higher order structure, including disulfide bonds |
| Denaturing | + | - | Higher order structure is disrupted, but disulfide bonds are retained |
| Reducing | - | + | Disulfides are removed, but most higher order structure remains |
| Native | - | - | The protein retains higher order structure. Multimers and protein-protein interactions can be detected |
Controls and Standards
It can be very useful to include a positive and a negative control on the gel along with the samples that are being evaluated. For a positive control it is typical to use a known source of target protein, such as purified protein or a control lysate, under conditions where it will be detectible by the antibody used in the experiment.
The positive control is important for confirming the identity of the target since it will produce a reference band on the blot showing the expected migration of the target protein and confirming the activity of the antibodies. Positive controls are also helpful for troubleshooting, and for comparing the data between separate blots.
Premade cellular and tissue lysates are now commercially available from a number of suppliers, including Bio-Rad, and can be used as a convenient positive control. In addition, commercially supplied tissue and cell lysates are suitable for troubleshooting protocols.
If possible, it is also helpful to include a negative sample control, such as known null cell line, as a means of confirming that the signal is specific to the desired protein.
The final component required for the gel is a molecular weight standard since a key feature of Western blotting is to provide information on the size of the protein. Also known as molecular weight markers, these are premade mixtures of proteins with known molecular weights, usually 5-6 proteins spanning the range from 10 kDa to 200 kDa.
Molecular weight standards come in a variety of formats, including unstained, prestained, multi-colored, or directly labeled for Western detection. They are an excellent means of monitoring progress while the gel is running, of checking transfer efficiency, and for orienting the immunoblot. Care should be taken not to overload the standards since they may obscure signal from the target protein .
Popular Resources

Generate Western Blot Data You Can Trust
Best practices for image documentation, integrity, and verification.
TidyBlot Western Blot Detection Reagent:HRP
TidyBlot Reagent:HRP is a western blotting detection reagent that specifically binds to native (non-reduced) antibodies.